
SSE #173: ESTRATÉGIAS NUTRICIONAIS PARA MELHORAR O CONTEÚDO E FUNÇÃO MITOCONDRIAL
Publicado em
November 2017
Autor
Grahm P. Holloway, PhD
PONTOS-CHAVE:
- O aumento do conteúdo mitocondrial induzido pelos treinamentos, melhora a tolerância aos exercícios por atenuar a elevação nas concentrações de adenosina difosfato (ADP) livre no citosol.
- As estratégias nutricionais para melhorar a biogênese mitocondrial induzida pelo exercício são limitadas, parcialmente devido à falta de entendimento sobre os sinais moleculares iniciais, que regulam este processo.
- Uma informação recente de que as espécies reativas de oxigênio (EROs) derivadas da mitocôndria podem induzir a biogênese mitocondrial, pode resultar em novas abordagens em relação aos treinamentos.
- Foi demonstrado que treinar em um “estado low carb”, aumentou o conteúdo mitocondrial, apesar do(s) mecanismo(s) responsável(is) por essa adaptação permanecer discutível.
- A ingestão do suco de beterraba (nitrato) não altera a eficiência de acoplamento mitocondrial, mas eleva as taxas de emissão das EROs mitocondriais, apesar da relevância biológica desta observação permanecer desconhecida.
- A resposta intrínseca da mitocôndria ao ADP é influenciada pelo exercício agudo e crônico, assim como pelo consumo de ácidos graxos poli-insaturados, portanto, a sensibilidade mitocondrial ao ADP pode ser alterada independentemente do conteúdo mitocondrial.
INTRODUÇÃO
Exercícios extenuantes podem aumentar as demandas energéticas da musculatura esquelética em 100 vezes além das necessidades de repouso, apresentando um enorme desafio às vias bioenergéticas para a manutenção das concentrações de adenosina trifosfato (ATP), a unidade básica de energia dentro do músculo. A performance do exercício é influenciada por diversos fatores, incluindo o fluxo sanguíneo, difusão de substratos metabólicos, metabolismo dentro da musculatura esquelética, e a habilidade em gerar força mecânica ideal/desejada. Enquanto a musculatura esquelética é equipada com uma série intrínseca de reações enzimáticas que ressintetizam ATP para garantir a sobrevivência celular durante estas condições, acredita-se que as mitocôndrias representem uma organela-chave, influenciando a homeostase metabólica na musculatura. O transporte de adenosina difosfato (ADP) do citoplasma para a matriz mitocondrial pode influenciar indiretamente o fluxo glicolítico (o ADP é um ativador alostérico das enzimas limitantes das taxas deste fluxo), e diretamente afetar as taxas de fosforilação oxidativa. Obtém-se como resultado, a melhora na performance do exercício, a economia de glicogênio muscular, a produção atenuada de lactato, e o aumento da confiança no metabolismo aeróbico após o treinamento, fatores que têm sido atribuídos a uma melhora na sensibilidade mitocondrial ao ADP, devido ao aumento no conteúdo mitocondrial (Holloszy & Coyle, 1984). Historicamente, esta resposta tem sido reconhecida integralmente pela indução da biogênese mitocondrial e ao maior conteúdo mitocondrial (Holloszy & Coyle, 1984); no entanto, a regulação externa das proteínas envolvidas no transporte mitocondrial de ADP (alteração na “eficiência”), provavelmente também existe. Este artigo do Sports Science Exchange (SSE) irá focar em discutir possíveis estratégias para aumentar 1) o conteúdo mitocondrial, ou 2) a eficiência mitocondrial. A consequência biológica em se aumentar o conteúdo ou a função mitocondrial, é uma melhora na sensibilidade ao ADP (como se discutirá a seguir), portanto, esta revisão também irá discutir 3) estratégias nutricionais e de treinamentos para melhorar diretamente a sensibilidade mitocondrial ao ADP.
A BIOGÊNESE MITOCONDRIAL
Já se conhece, por quase um século, que os atletas de elite têm uma maior taxa máxima de consumo de oxigênio (VO2max) e maior atividade enzimática mitocondrial, no final, contribuindo com performances de elite. Enquanto isto foi originalmente atribuído à genética, a clássica pesquisa de Holloszy, em 1967, demonstrou a impressionante plasticidade da musculatura esquelética em relação ao aumento do conteúdo mitocondrial e a melhora na capacidade de exercício (Holloszy, 1967). Este trabalho de referência descreveu a observação básica de que treinos com cargas excessivas aumentam o conteúdo mitocondrial, mas não alteram as funções intrínsecas da mitocôndria. Como resultado, as pesquisas pelos próximos 50 anos focaram em elucidar os mecanismos responsáveis pela indução da biogênese mitocondrial. Esta revisão não irá focar em uma descrição detalhada dos processos que resultam na indução da biogênese mitocondrial, mas uma breve descrição é necessária para fornecer um esquema básico para a discussão sobre as estratégias, com o objetivo de otimizar esta resposta.
O proteoma mitocondrial consiste em ~1.600 proteínas, a grande maioria sendo codificada no núcleo, já que o DNA mitocondrial (DNAmt) apenas transcreve 13 subunidades de proteína da cadeia transportadora de elétrons e a proteína necessária para a translocação de RNAm dentro desta organela (para uma revisão, veja Bartlett et al., 2015). A indução da biogênese mitocondrial, portanto, envolve uma resposta de sinalização coordenada que estimula ambos os genomas. A identificação da proteína proliferadora de peroxissoma com receptor ɣ co-ativador 1α (PGC-1α) como um co-ativador transcricional que sincroniza este processo, foi o principal avanço no entendimento sobre os mecanismos moleculares que regulam o conteúdo mitocondrial. A ativação citoplasmática induzida pelo cálcio da proteína quinase dependente de Ca2+/calmodulina (CaMK), a ativação da quinase por adenosina monofosfato (AMPK) pelo turnover de energia, e a maior produção das espécies reativas de oxigênio (EROs), têm todas sido implicadas como mecanismo primário na indução da biogênese mitocondrial (Bartlett et al., 2015). No entanto, enquanto os pesquisadores continuam a melhorar o nosso entendimento sobre os processos envolvidos no aumento do volume mitocondrial, nosso conhecimento sobre os sinais que iniciam a biogênese mitocondrial permanece não muito bem definido, limitando a nossa habilidade em desenvolver intervenções ideais para os treinamentos.
Apesar da limitação no nosso entendimento molecular sobre a biogênese mitocondrial, estratégias de treinamento que aumentam estas respostas têm sido identificadas. De particular interesse é a noção sobre a periodização de treinos em “estado low carb”, uma abordagem que tem sido mostrada: 1) ativar as vias moleculares associadas com a biogênese mitocondrial, 2) aumentar a capacidade oxidativa da musculatura, e em algumas situações 3) melhorar a capacidade de exercício (Bartlett et al., 2015). O trabalho de referência de Pilegaard e colaboradores foi fundamental em destacar que a baixa disponibilidade de glicogênio, durante e após o exercício, amplifica a expressão normal dos genes mitocondriais, induzida pelo exercício (Pilegaard et al., 2002; 2005). Por outro lado, outros trabalhos têm mostrado que o exercício agudo na presença de grande disponibilidade de carboidratos atenua os sinais associados com a biogênese mitocondrial (Bartlett et al., 2013). Principalmente, estes achados sobre os sinais agudos transitórios em indivíduos não treinados parecem ser traduzidos aos atletas, já que a periodização de treinos em “estado” de baixa concentração de glicogênio, de forma semelhante aumenta o conteúdo mitocondrial em indivíduos altamente treinados. Especificamente, o grupo de Hawley demonstrou que o treino duas vezes ao dia, a cada dois dias, em indivíduos já treinados, aumenta o conteúdo de glicogênio muscular, marcadores de conteúdo mitocondrial, e as taxas de oxidação de gordura, enquanto que a mesma quantidade de trabalhos que separou sessões únicas de exercício em dias consecutivos, não demonstrou estes resultados (Yeo et al., 2008). Adicionalmente, a privação de carboidratos após uma sessão de treinamento a noite mostrou uma melhora nos tempos de corrida de 10 km (Marquet et al., 2016), sugerindo que possíveis benefícios na performance estão associados com as respostas moleculares observadas. Estas adaptações benéficas observadas com a periodização dos treinos em “estado low carb” temporário foram atribuídas a ativação da AMPK (Yeo et al., 2008), já que trabalhos anteriores destacaram uma propriedade de ligação do glicogênio em subunidades β da AMPK e ativação na presença de baixo conteúdo de glicogênio dentro do músculo (McBride et al., 2009; Wojtaszewski et al., 2003). No entanto, enquanto grande parte da literatura se dedicou em estudar o papel do turnover energético e ativação da AMPK como sinal essencial para a indução da biogênese mitocondrial (revisado em Marcinko & Steinberg, 2014), modelos genéticos que apresentam a atividade de APMK substancialmente prejudicada foram confundidos pelos possíveis danos da performance cardiovascular durante o exercício, tornando as interpretações mais difíceis. Em contraste, a neutralização da quinase hepática B1 (LKB1), um ativador da AMPK na musculatura de roedores, prejudica a capacidade de exercício e reduz o conteúdo mitocondrial em animais sedentários, mas não afeta as respostas aos exercícios (Tanner et al., 2013). Isto sugere que a ativação da AMPK não é necessária para a indução da biogênese mitocondrial. Portanto, enquanto o treinamento em um “estado low carb” parece aumentar o conteúdo mitocondrial, o mecanismo molecular permanece discutível.
As EROs também foram consideradas como um sinal para a indução da biogênese mitocondrial, mas evidências claras para um papel mecânico das EROs não foram previamente estabelecidas. No entanto, um argumento teórico foi realizado baseado nas observações que os exercícios aumentam o dano oxidativo da musculatura (Davies et al., 1982). E ainda, diversas linhas de evidência foram recentemente estabelecidas para implicar as EROs, e especificamente as EROs derivadas da mitocôndria, na indução da biogênese mitocondrial. Especificamente, o consumo de dietas ricas em gordura demonstram aumentar o conteúdo mitocondrial (Jain et al., 2014), a emissão mitocondrial de EROs, as alterações de oxido-redução (redox) no canal de liberação de cálcio receptores de rianodina (RyR – sigla em inglês), e a ativação da sinalização de cálcio (CaMK II) (Jain et al., 2014). Estas respostas foram inteiramente prevenidas com o consumo de antioxidantes direcionados às mitocôndrias (SkQ) (Jain et al., 2014). Além disso, foi demonstrado que uma única sessão de treino intervalado de alta intensidade aumenta a fragmentação mediada pelas EROs da RyR em associação com a indução da biogênese mitocondrial (Place et al., 2015). Estas respostas foram atenuadas após o treino crônico, possivelmente explicando o rendimento reduzido nos exercícios, em relação à expansão constante no volume mitocondrial (Place et al., 2015). Em conjunto, estes resultados sugerem que as EROs derivadas da mitocôndria são um sinalizador molecular importante para a indução da biogênese mitocondrial, um processo que pode necessitar de modificações redox da sinalização mediada pela RyR e cálcio. Estes dados ajudam a explicar a falta da biogênese mitocondrial observada em indivíduos consumindo altas quantidades de certos antioxidantes enquanto se exercitam (Paulsen et al., 2014). Desta maneira, aumentar o estresse redox durante o exercício/recuperação pode aumentar a biogênese mitocondrial. Enquanto apenas uma especulação, um treino em “estado low carb” pode promover a sinalização redox, já que os ácidos graxos têm alta propensão em produzir as EROs derivadas da mitocôndria. Claramente, pesquisas futuras são necessárias para delinear totalmente o papel da sinalização redox na indução da biogênese mitocondrial, e para estabelecer novos paradigmas de treinamentos para maximizar estes processos em atletas.
SENSIBILIDADE MITOCONDRIAL AO ADP
A indução da biogênese mitocondrial, e a melhora consecutiva na sensibilidade mitocondrial ao ADP se tornou sinônimo das adaptações ao treinamento. Enquanto diversos processos poderiam influenciar as concentrações livres de ADP in vivo, avaliações diretas da respiração mitocondrial utilizando fibras musculares permeabilizadas têm consistentemente mostrado 1) melhora na respiração submáxima, ou 2), por outro lado, uma redução na quantidade necessária de ADP para manter um determinado fluxo aeróbico. Estes achados sugerem que alterações mitocondriais contribuem com a melhora na sensibilidade ao ADP após o treinamento (Figura 1A, B). Este modelo clássico de trabalho é baseado na crença de que a “função” mitocondrial permanece inalterada após intervenção com treinamento crônico. No entanto, evidências estão surgindo e sugerindo que o transporte mitocondrial de ADP é um processo regulado, aumentando a possibilidade de que intervenções relacionadas ao estilo de vida podem influenciar a sensibilidade mitocondrial ao ADP, na ausência de maior conteúdo mitocondrial. Paradoxalmente, a concentração necessária de ADP para permitir a respiração meio-máxima (Km), denominada de Km aparente de ADP, é reduzida após o treinamento (Figura 1C, D), sugerindo que a sensibilidade intrínseca de um determinado mitocondrion ao ADP é atenuada com o treinamento. Enquanto esta definição bioquímica não tem relevância biológica direta, ela demonstra que a sensibilidade ao ADP pode ser regulada externamente, e um maior entendimento da regulação deste processo pode gerar alguns insights sobre novos programas de treinamentos.

Figura 1. Estes esquemas ilustram a resposta da cinética respiratória da adenosina difosfato (ADP) em fibras musculares permeabilizadas, antes e após o treino. Nestes experimentos piruvato saturado mais malato foram fornecidos, e depois consecutivamente, o ADP é inserido em um sistema selado para medir a condução respiratória. Se estes valores são expressos em valores absolutos (normalizados com o peso seco como em A e B) o aumento na respiração máxima reflete a biogênese mitocondrial e um aumento no conteúdo mitocondrial. Como resultado, a quantidade necessária de ADP para atingir uma taxa específica do consumo de oxigênio foi menor após o treino. No entanto, se o conteúdo mitocondrial é impedido em expressar a respiração como um percentual máximo do consumo de oxigênio (C e D), uma observação diferente é evidente. Especificamente, após o treino uma concentração maior de ADP foi necessária para atingir 50% da respiração máxima (denominada de Km aparente de ADP). Estas últimas análises sugerem que a sensibilidade intrínseca da mitocôndria foi na verdade reduzida com o treino. JO2, taxa de consumo de oxigênio ou respiração; Vmax, taxa máxima de atividade.
Tradução da figura:
JO2 é a taxa de consumo de oxigênio, como exemplo a respiração mitocondrial
A (gráfico JO2 pmol/seg/mg/peso seco)
Vmax maior após o treino por causa do maior conteúdo mitocondrial
(Pós-treino/Pré-treino)
B (gráfico JO2 pmol/seg/mg/peso seco)
O eixo “x” redimensionado destaca que menor quantidade de ADP é necessária para desencadear uma determinada taxa de consumo de oxigênio após o treino
(Pós-treino/Pré-treino)
C (gráfico JO2 (% máxima da respiração)
Os mesmos dados expressos em relação a Vmax destaca que sensibilidade intrínseca da mitocôndria ao ADP é atenuada com o treino
(Pós-treino/Pré-treino)
D (gráfico JO2 (% máxima da respiração)//concentração de ADP (µmol)
O eixo “x” redimensionado destaca que maior quantidade de ADP é necessária para desencadear 50% do consumo máximo de oxigênio após o treino
(Pós-treino/Pré-treino)
Enquanto a adenina nucleotídeo translocase (ANT – sigla em inglês) é necessária para a troca de ADP/ATP (Figura 2), acredita-se que a creatina quinase mitocondrial (CKmit) concentre o ADP no espaço intermembranas para otimizar a difusão do ADP na mitocôndria, enquanto a fosfocreatina (PCr)/Creatina (Cr) é estimulada a sofrer difusão ~2.000 vezes mais rápida através da membrana mitocondrial externa/através do citoplasma (Wallimann et al., 2011). Portanto, acredita-se que a transferência de fosfatos através das reações de creatina quinase contribuem substancialmente para a homeostase metabólica, particularmente durante a contração muscular onde as necessidades de ATP podem aumentar ~100 vezes (Saks et al., 1985). Contudo, a ANT é necessária para ambos os transportes de ADP/ATP dependente e independente de Cr, através da membrana mitocondrial interna, e a regulação externa da ANT existe, já que uma sessão aguda de exercícios intervalados de alta intensidade demonstrou melhorar de maneira aguda a sensibilidade mitocondrial ao ADP (Ydfors et al., 2016), enquanto exercícios constantes (~60% do VO2max por duas horas) atenuaram a sensibilidade mitocondrial ao ADP na ausência de Cr (Perry et al., 2012). Estes dados sugerem que a regulação da ANT é altamente complexa e dependente da intensidade do exercício (Figura 2). Enquanto o nosso entendimento sobre a regulação da sensibilidade mitocondrial ao ADP é incompleto, sabe-se que o ácido graxo palmitoil-CoA interage com a ANT para inibir a troca de ADP/ATP, um processo atenuado pelos treinamentos crônicos (Ludzki et al., 2015), que em teoria poderia contribuir para “tamponar os aumentos de ADP citoplasmático livre” durante exercícios após o treino. Claramente, cada vez mais, as evidências têm apoiado a sugestão de que a sensibilidade mitocondrial ao ADP é regulada extensivamente durante o exercício agudo e é influenciada pelo treinamento crônico (Figura 2). Em conjunto, estes dados destacam a possibilidade de alteração na sensibilidade mitocondrial ao ADP, na ausência da biogênese mitocondrial, desafiando o preceito sustentado por tanto tempo de que aumentos no conteúdo mitocondrial são necessários para a alteração da fonte energética e melhora na performance.
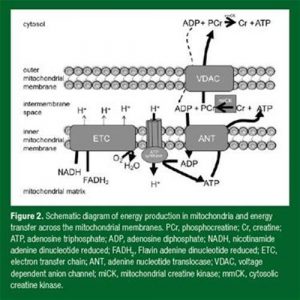
Figura 2. Diagrama da produção de energia na mitocôndria e transferência de energia pela membrana mitocondrial. PCr, fosfocreatina; Cr, creatina; ATP, adenosina trifosfato; ADP, adenosina difosfato; NADH, nicotinamida adenina dinucleotídeo reduzida; FADH2 , flavina adenina dinucleotídeo reduzida; ETC, cadeia transportadora de elétrons; ANT, adenina nucleotídeo translocase; VDAC, canais de ânion dependente de voltagem; miCK, creatina quinase mitocondrial; mmCK, creatina quinase citosólica.
Tradução da figura:
Citosol
Membrana mitocondrial externa
Espaço intermembranas
Membrana mitocondrial interna
Matriz mitocondrial
ÁCIDOS POLI-INSATURADOS E A BIOENERGÉTICA MITOCONDRIAL
Dada a relativa novidade de identificar do transporte mitocondrial de ADP como um processo regulado, muito poucas evidências foram geradas em relação às abordagens nutricionais para aumentar este processo. No entanto, a suplementação com o ácido eicosapentaenóico (EPA) e o ácido docosahexaenóico (DHA) foi mostrada alterar a composição lipídica das membranas mitocondriais em associação com um aumento na sensibilidade mitocondrial ao ADP (Herbst et al., 2014). Especificamente, uma suplementação diária de 2 g de EPA e 1g de DHA em indivíduos saudáveis (média de idade de 22 anos) melhorou a sensibilidade da mitocôndria ao ADP, em fibras musculares permeabilizadas, em ~30% na ausência de alterações no conteúdo mitocondrial (Herbst et al., 2014). Curiosamente, a oferta de EPA/DHA também aumentou as taxas mitocondriais de emissão das EROs (Herbst et al., 2014), e apesar disto não ter resultado na indução da biogênese mitocondrial em indivíduos sedentários, aumentou o potencial desta abordagem nutricional para melhorar a biogênese mitocondrial induzida pelo exercício. No entanto, já que a suplementação com EPA/DHA foi relacionada com a melhora na síntese de proteínas, performance cognitiva, função imunológica, integridade óssea, função cardiovascular e a expressão de genes associados com a oxidação lipídica em diversos tecidos (revisada em Jeromson et al., 2015), é tentador especular que a suplementação com EPA/DHA poderia melhorar a performance do exercício. Contudo, há uma escassez na literatura relacionada a habilidade do EPA/DHA em melhorar as respostas metabólicas durante o exercício na musculatura esquelética humana.
A BIOENERGÉTICA MITOCONDRIAL E DO NITRATO
Os princípios tradicionais estabelecem que a função mitocondrial não é externamente regulada além da provisão de substratos necessários para a fosforilação oxidativa. Esta crença vai além da observação original de Holloszy de que avaliações estequiométricas mitocondriais in vitro (razão P/O: o ADP consumido por átomo de oxigênio) permanecem constantes após treinos crônicos (Holloszy, 1967). No entanto, os achados de referência de Larsen e colaboradores mostraram que a ingestão de nitrato de sódio da dieta, por 3 dias (consumo diário de ~7mmol de nitrato de sódio), melhorou a eficiência de acoplamento mitocondrial, as taxas máximas de produção de ATP, e reduziu o consumo de oxigênio em todo corpo humano (Larsen et al., 2011), indicaram que esta noção precisava ser reconsiderada. Curiosamente, o consumo oral de suco de beterraba também diminui o gasto de oxigênio nos exercícios submáximos em humanos, sugerindo que as fontes orais de nitrato melhoram universalmente a eficiência respiratória mitocondrial. Contudo, em contraste com o nitrato de sódio, consumir uma quantidade maior de nitrato oralmente na forma de suco de beterraba (~26mmol de nitrato diariamente), por sete dias, não altera a proporção isolada de acoplamento mitocondrial, a respiração espontânea, o potencial de membrana mitocondrial ou a sensibilidade mitocondrial ao ADP em fibras musculares permeabilizadas (Whitfield et al., 2015), indicando que o suco de beterraba não altera a eficiência de acoplamento mitocondrial. Estas observações “in vitro” têm suporte dos achados de que as taxas de ressíntese de PCr in vivo, um metabolismo oxidativo mitocondrial estimado, não foram alteradas após o consumo de suco de beterraba por seis dias, mas ao invés disso, as taxas de hidrólise de ATP foram reduzidas (Bailey et al., 2010). Em conjunto, estes dados sugerem que o mecanismo de ação do suco de beterraba não envolve uma melhora nas taxas de eficiência mitocondrial, mas sim uma melhora na eficiência mecânica. Além disso, a oferta de nitrato de sódio reduziu o conteúdo de proteína ANT (Larsen et al., 2011), e enquanto isto pode melhorar as taxas de acoplamento mitocondrial, seria esperado reduzir a sensibilidade mitocondrial ao ADP , o que seria contraprodutivo em relação às respostas aos treinamentos (Figura 2).
Enquanto o suco de beterraba não parece influenciar o metabolismo oxidativo mitocondrial, tem sido mostrado que aumenta as taxas de emissão de EROs mitocondriais (Whitfield et al., 2015), o que poderia contribuir com a aparente melhora na eficiência do exercício após o consumo de suco de beterraba. Enquanto uma relação direta de causa e efeito entre as EROs mitocondriais e uma melhora na eficiência mecânica da musculatura permanece a ser determinada, os mecanismos pertencentes as modificações redox nos sarcômeros (exemplo, troponina I) e a condução do cálcio (exemplo, RyR e retículo sarco/endoplasmático Ca2+-ATPase [SERCA – sigla em inglês]) permanecem como possíveis fatores (Hernandez et al., 2012). A necessidade das EROs mitocondriais na mediação da melhora na performance do exercício com o suco de beterraba é um modelo atraente, dada a conhecida redução das EROs mitocondriais após o treinamento (Place et al., 2015), e a aparente resistência/atenuação nas respostas ao suco de beterraba em atletas de resistência de elite (Boorsma et al., 2014). Também é tentador especular que o consumo do suco de beterraba durante programas crônicos de exercícios iria aumentar a biogênese mitocondrial como resultado de um aumento na transcrição genética mediada pelas EROs mitocondriais; no entanto, esta possibilidade aguarda respaldo científico. Parece que o suco de beterraba não altera a eficiência de acoplamento mitocondrial (Bailey et al., 2010; Whitfield et al., 2015). Este achado de referência de que o nitrato de sódio melhora a eficiência de acoplamento mitocondrial aumenta a possibilidade para que futuros alvos nutricionais sejam identificados com efeitos biológicos similares (Larsen et al., 2011).
RESUMO E APLICAÇÕES PRÁTICAS
Melhorar o conteúdo e/ou função mitocondrial seria vantajoso para a performance do exercício, e portanto, um entendimento básico da regulação da mitocôndria é necessário para elucidar novas abordagens para modular esta organela dinâmica. A nova ligação mecânica entre a emissão das EROs mitocondriais e a biogênese mitocondrial aumenta a possibilidade de inúmeras abordagens nutricionais a serem testadas em conjunto com programas de treinamentos em atletas. Em particular, o consumo de EPA/DHA e suco de beterraba foi mostrado que aumentam as taxas de emissão das EROs mitocondriais. A relevância biológica desta observação permanece desconhecida, mas pode contribuir com as conhecidas melhoras na eficiência mecânica e redução no consumo de oxigênio observados com a suplementação de suco de beterraba. Há claras evidências de que treinar em “estado low carb” aumenta o conteúdo mitocondrial, e seria intrigante determinar se o consumo do suco de beterraba poderia aumentar esta resposta. O maior benefício do suco de beterraba é a natureza imediata deste suplemento (exemplo, horas e dias), enquanto em contraste a limitação de EPA/DHA está na necessidade do consumo crônico destes lipídeos (exemplo, semanas e meses), que também tem sido mostrado atenuar os sinais associados com a síntese de proteínas.
Este efeito biológico no aumento do conteúdo mitocondrial é uma melhora na sensibilidade mitocondrial ao ADP . Curiosamente, exercícios intermitentes de alta intensidade demonstraram melhorar de maneira aguda a sensibilidade mitocondrial ao ADP , aumentando a possibilidade de sessões breves de exercícios com alta intensidade no período de aquecimento, podendo melhorar o controle metabólico na sessão subsequente de exercícios. Isto pode ser particularmente benéfico aos atletas, já que o treinamento reduz a sensibilidade intrínseca mitocondrial ao ADP (veja Figuras 1C, D). Contudo, o tempo mínimo e intensidade do exercício para atingir esta melhora na sensibilidade ao ADP precisam ser determinados para garantir que as depleções do glicogênio muscular não ocorram imediatamente antes de competições, já que isto pode ser contraprodutivo. O recente avanço no nosso entendimento sobre a regulação do transporte mitocondrial de ADP elucidou lacunas nos nossos modelos de trabalho que precisam ser abordadas. No entanto, a consciência destas lacunas no nosso conhecimento cria a possibilidade para abordagens experimentais únicas a serem desenhadas com o objetivo de melhorar a performance do exercício no futuro.
REFERÊNCIAS
Bailey, S.J., J. Fulford, A. Vanhatalo, P.G. Winyard, J.R. Blackwell, F.J. DiMenna, D.P. Wilkerson, N. Benjamin, and A.M. Jones (2010). Dietary nitrate supplementation enhances muscle contractile efficiency during knee-extensor exercise in humans. J. Appl. Physiol. 109:135-148.
Bartlett, J.D., J.A. Hawley, and J.P. Morton (2015). Carbohydrate availability and exercise training adaptation: too much of a good thing? Eur. J. Sport Sci. 15:3-12.
Bartlett, J.D., J. Louhelainen, Z. Iqbal, A.J. Cochran, M.J. Gibala, W. Gregson, G.L. Close, B. Drust, and J. P. Morton (2013). Reduced carbohydrate availability enhances exercise-induced p53 signaling in human skeletal muscle: implications for mitochondrial biogenesis. Am. J. Physiol. 304, R450-R458.
Boorsma, R.K., J. Whitfield, and L.L. Spriet (2014). Beetroot juice supplementation does not improve running performance in 1500 m runners. Med. Sci. Sports Exerc. 46:2326-2334.
Davies, K.J., A.T. Quintanilha, G. A Brooks, and L. Packer (1982). Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 107:1198-1205.
Herbst, E.A., S. Paglialunga, C. Gerling, J. Whitfield, K. Mukai, A. Chabowski, G.J. Heigenhauser, L.L. Spriet, and G.P. Holloway (2014). Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J. Physiol. 592:1341-1352.
Hernandez, A., T.A. Schiffer, N. Ivarsson, A.J. Cheng, J.D. Bruton, J.O. Lundberg, E. Weitzberg, and H. Westerblad (2012). Dietary nitrate increases tetanic [Ca2+]i and contractile force in mouse fast-twitch muscle. J. Physiol. 590:3575-3583.
Holloszy, J.O. (1967). Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 242:2278-2282.
Holloszy, J.O., and E.F. Coyle (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. 56:831-838.
Jain, S.S., S. Paglialunga, C. Vigna, A. Ludzki, E.A. Herbst, J.S. Lally, P. Schrauwen, J. Hoeks, A.R. Tupling, A. Bonen, and G.P. Holloway (2014). High-fat diet-induced mitochondrial biogenesis is regulated by mitochondrial-derived reactive oxygen species activation of CaMKII. Diabetes 63:1907-1913.
Jeromson, S., I.J. Gallagher, S.D. Galloway, and D.L. Hamilton (2015). Omega-3 fatty acids and skeletal muscle health. Mar. Drugs 13:6977-7004.
Larsen, F.J., T.A. Schiffer, S. Borniquel, K. Sahlin, B. Ekblom, L.O. Lundberg, and E. Weitzberg (2011). Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 13:149-159.
Ludzki, A., S. Paglialunga, B.K. Smith, E.A. Herbst, M.K. Allison, G.J. Heigenhauser, P.D. Neufer, and G.P. Holloway (2015). Rapid repression of ADP transport by palmitoyl-CoA is attenuated by exercise training in humans: A potential mechanism to decrease oxidative stress and improve skeletal muscle insulin signaling. Diabetes 64:2769-2779.
Marcinko, K., and G.R. Steinberg (2014). The role of AMPK in controlling metabolism and mitochondrial biogenesis during exercise. Exp. Physiol. 99:1581-1585.
Marquet, L.A., J. Brisswalter, J. Louis, E. Tiollier, L.M. Burke, J.A. Hawley, and C. Hausswirth (2016). Enhanced endurance performance by periodization of carbohydrate intake: “Sleep low” strategy. Med. Sci. Sports Exerc. 48:663-672.
McBride, A., S. Ghilagaber, A. Nikolaev, and D.G. Hardie (2009). The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 9:23-34.
Paulsen, G., K.T. Cumming, G. Holden, J. Hallen, B.R. Ronnestad, O. Sveen, A. Skaug, I. Paur, N.E. Bastani, H.N. Ostgaard, C. Buer, M. Midttun, F. Freuchen, H. Wiig, E.T. Ulseth, I. Garthe, R. Blomhoff, H.B. Benestad, and T. Raastad (2014). Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double-blind, randomised, controlled trial. J. Physiol. 592:1887-1901.
Perry, C.G., D.A. Kane, E.A. Herbst, K. Mukai, D.S. Lark, D.C. Wright, G.J. Heigenhauser, P.D. Neufer, L.L. Spriet, and G.P. Holloway (2012). Mitochondrial creatine kinase activity and phosphate shuttling are acutely regulated by exercise in human skeletal muscle. J. Physiol. 590:5475-5486.
Pilegaard, H., C. Keller, A. Steensberg, J.W. Helge, B.K. Pedersen, B. Saltin, and P.D. Neufer (2002). Influence of pre-exercise muscle glycogen content on exercise-induced transcriptional regulation of metabolic genes. J. Physiol. 541:261-271.
Pilegaard, H., T. Osada, L.T. Andersen, J.W. Helge, B. Saltin, and P.D. Neufer (2005). Substrate availability and transcriptional regulation of metabolic genes in human skeletal muscle during recovery from exercise. Metabolism 54:1048-1055.
Place, N., N. Ivarsson, T. Venckunas, D. Neyroud, M. Brazaitis, A.J. Cheng, J. Ochala, S. Kamandulis, S. Girard, G. Volungevicius, H. Pauzas, A. Mekideche, B. Kayser, V. Martinez-Redondo, J.L. Ruas, J. Bruton, A. Truffert, J.T. Lanner, A. Skurvydas, and H. Westerblad (2015). Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high-intensity interval exercise. Proc. Natl. Acad. Sci. 112:15492-15497.
Saks, V.A., A.V. Kuznetsov, V.V. Kupriyanov, M.V. Miceli, and W.E. Jacobus (1985). Creatine kinase of rat heart mitochondria. The demonstration of functional coupling to oxidative phosphorylation in an inner membrane-matrix preparation. J. Biol. Chem. 260:7757-7764.
Tanner, C.B., S.R. Madsen, D.M. Hallowell, D.M. Goring, T.M. Moore, S.E. Hardman, M.R. Heninger, D.R. Atwood, and D.M. Thomson (2013). Mitochondrial and performance adaptations to exercise training in mice lacking skeletal muscle LKB1. Am. J. Physiol. 305:E1018-E1029.
Wallimann, T., M. Tokarska-Schlattner, and U. Schlattner (2011). The creatine kinase system and pleiotropic effects of creatine. Amino Acids 40:1271-1296.
Whitfield, J., A. Ludzki, G.J. Heigenhauser, J.M. Senden, L.B. Verdijk, L.J. van Loon, L.L. Spriet. and G.P. Holloway (2015). Beetroot juice supplementation reduces whole body oxygen consumption but does not improve indices of mitochondrial efficiency in human skeletal muscle. J. Physiol. 64:1419-1425.
Wojtaszewski, J.F., C. MacDonald, J.N. Nielsen, Y. Hellsten, D.G. Hardie, B.E. Kemp, B. Kiens, and E.A. Richter (2003). Regulation of 5’AMP-activated protein kinase activity and substrate utilization in exercising human skeletal muscle. Am. J. Physiol. 284:E813-822.
Ydfors, M., M.C. Hughes, R. Laham, U. Schlattner, J. Norrbom, and C.G. Perry (2016). Modelling in vivo creatine/phosphocreatine in vitro reveal divergent adaptations in human muscle mitochondrial respiratory control by ADP after acute and chronic exercise. J. Physiol. 594:3127-3140.
Yeo, W.K., C.D. Paton, A.P. Garnham, L.M. Burke, A.L. Carey, and J.A. Hawley (2008). Skeletal muscle adaptation and performance responses to once a day versus twice every second day endurance training regimens. J. Appl. Physiol. 105:1462-1470.
Baixar

Distribuir



